Biocatalytic platforms occupy a preferential position in the green chemistry agenda and the catalogue of natural enzymes allied with current
... moreBiocatalytic platforms occupy a preferential position in the green chemistry agenda and the catalogue of natural enzymes allied with current biotechnological knowledge offers a unique opportunity in this area. Despite the pleyades of Nature's enzymatic systems, its repertoire only covers a limited scope of the current need of chemical industries of catalytic processes. As a consequence, the discovery of
de novo enzymes which activity are absent in Nature is a field of research extremely active.
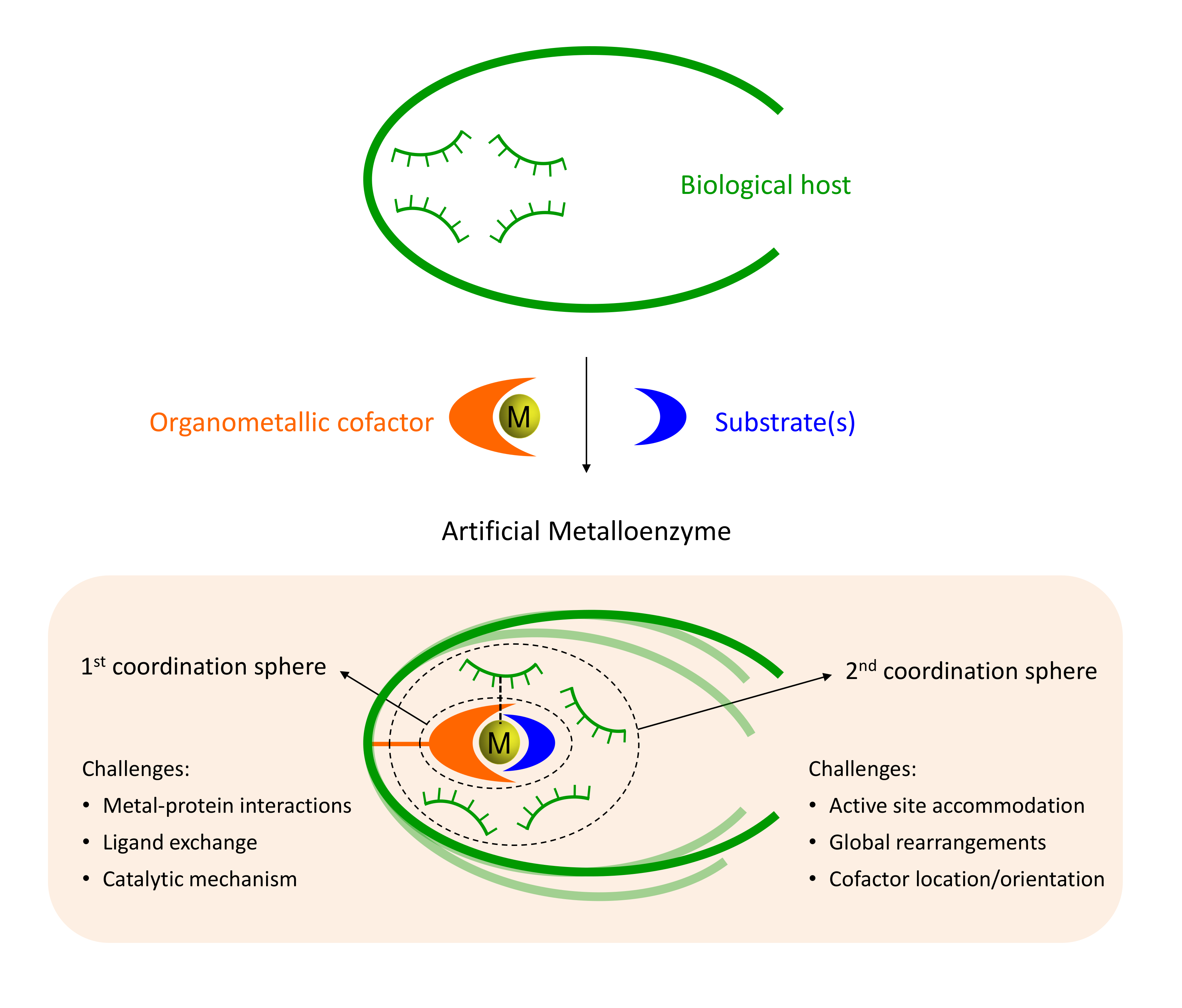
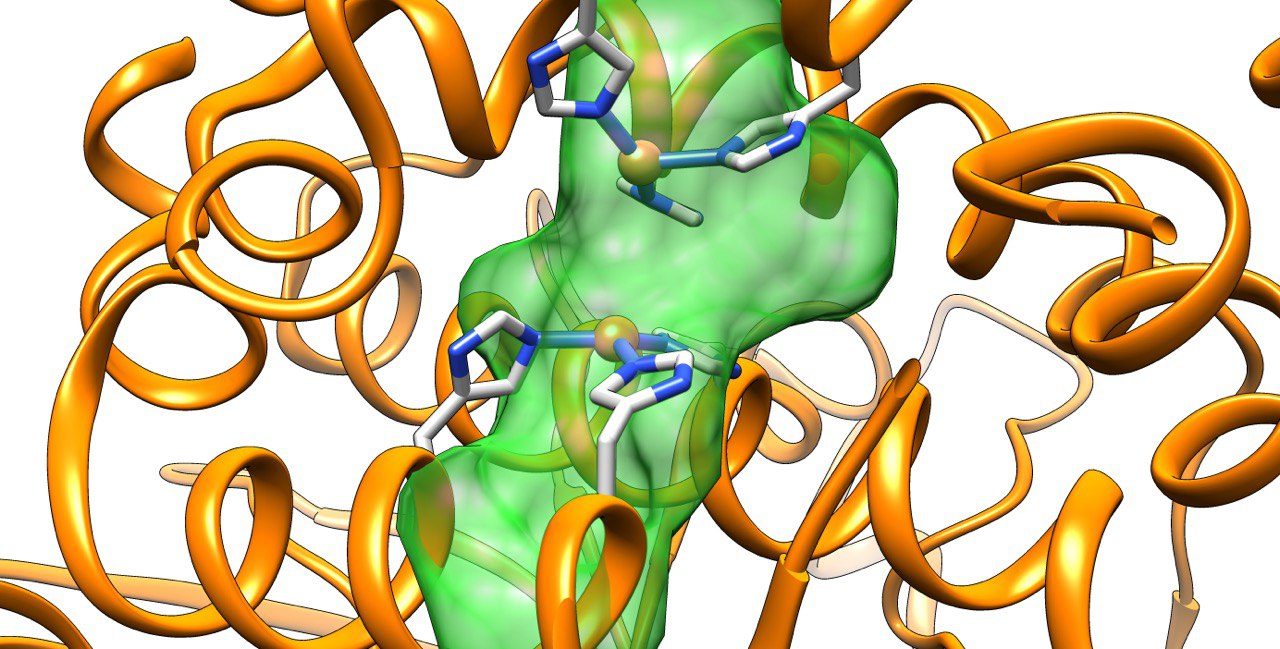
Amongst the most promising strategies are those based on the insertion of synthetic homogeneous catalyst into a biological scaffold. Somehow reminiscent of naturally occurying hemoenzymes, those systems provide many advantages for the future of biocatalysis. Howevever, the biohybrids designed to date still present many challenges like the identification the exact location of the cofactor, the characterization of catalytically consistent orientations of the substrate or to design variants with specific substrate, regio- or enantiospecificities.
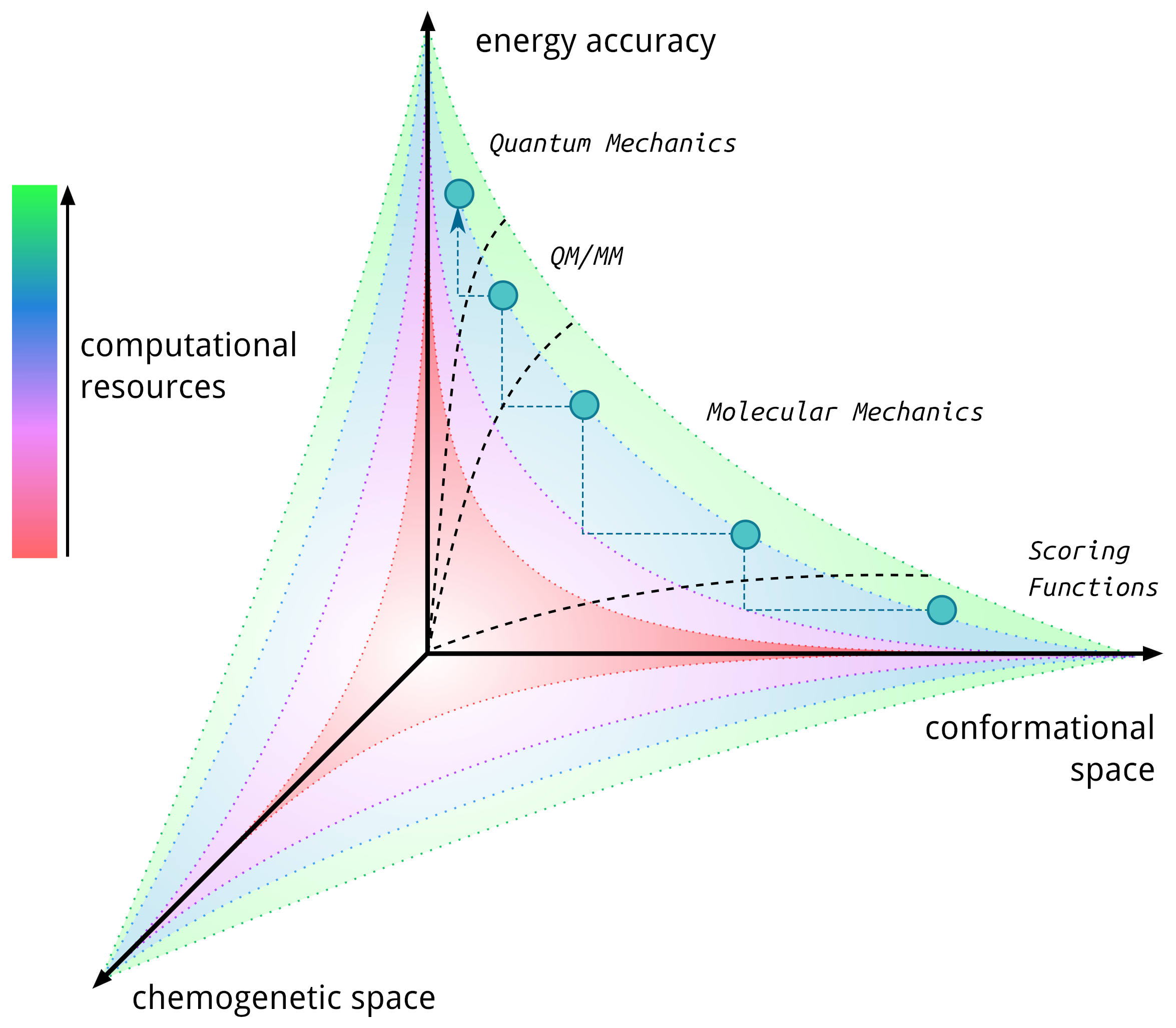
Our group has pioneered researches in the field of artificial metalloenzymes which relied on the development of computational integrative strategies including docking, MD and QM (QM/MM). We also tripled our efforts to update the computational framework with predictive tools for the binding of metallic species to protein scaffold. In particular, we are constantly developing new tools able to predict the binding of metallic compounds to biological hosts using structure based approaches and protein-ligand docking in a first instance.
We are lucky to enjoy collaborations with international leaders in the field like Profs. Thomas Ward (Basel), Gerard Roelfes (Groningen) and Jean-Pierre Mahy (Orsay).
Less